

Abstract
Patients with myelofibrosis who are treated with the Type I ATP competitive JAK inhibitor ruxolitinib derive symptomatic benefit from improvement in cytokine-mediated symptoms, and show cytoreductive responses in disease manifestations, such as splenomegaly or leukocytosis. However, ruxolitinib treatment does not consistently reduce JAK2 mutant allele burden nor eradicate the disease. Secondly, when ruxolitinib is withdrawn there is rapid recrudescence of cytokine-mediated symptoms within days of treatment stopping and in rare cases this has led to a life-threatening "ruxolitinib discontinuation syndrome" characterized by acute relapse of disease symptoms, splenomegaly, worsening cytopenia, and a cytokine storm akin to septic shock. Paradoxically, Type I inhibitors can induce the accumulation of JAK activation loop phosphorylation for unknown reasons, despite blockade of kinase function and inhibition of STAT phosphorylation. Here, we developed a time-dependent assay in primary myelofibrosis samples to mimic ruxolitinib withdrawal syndrome and investigate a possible mechanism of action involving JAK2 activation loop phosphorylation.
Methods: Primary peripheral blood de novo myelofibrosis samples were obtained prior to treatment with informed consent according to institutional guidelines (Stanford University Institutional Review Board No. 6453 or Board No. SACRB-APP-020). TF1.8 cells were cultured in RPMI with 10% fetal calf serum supplemented with 2 ng/ml of GM-CSF. SET-2 cells were cultured in RPMI with 10% serum. Phosphatase assays were performed with recombinant JAK2 kinase domain was mixed with recombinant PTP1B and JAK inhibitor. All antibodies were sourced from Cell Signalling Technologies. Unless otherwise stated, P values comparing two means were calculated using the two-tailed unpaired Student's t-test in Prism version 6 (GraphPad Software, Inc. La Jolla, CA).
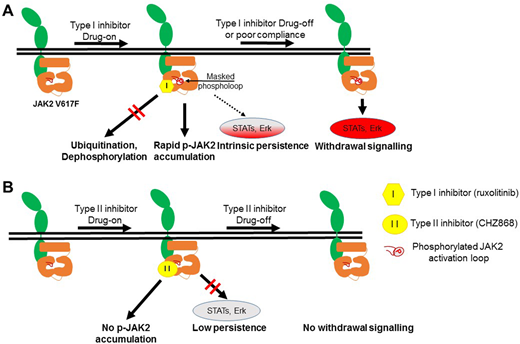
Results: We developed a time-dependent assay to mimic ruxolitinib withdrawal in primary JAK2V617F and CALR mutant myelofibrosis patient samples and observed striking activation of spontaneous STAT signaling in JAK2V617F samples after drug washout. Accumulation of ruxolitinib-induced loop phosphorylation was dose-dependent and correlated with rebound signaling and the presence of a JAK2V617F mutation. Ruxolitinib prevented dephosphorylation of a cryptic site involving Tyr1007/1008 in JAK2 blocking ubiquitination and degradation. We note that one patient with CALR mutant did not exhibit the same degree of STAT spontaneous withdrawal signalling compared to JAK2 V617F. Similarly, CALR mutant myelofibrosis cells showed an undetectable level of activated JAK2 Tyr1007/1008 phosphorylation in the presence of ruxolitinib. This is consistent with a number of emerging reports using CALR mutated models of myelofibrosis but needs to be confirmed in further patient samples.
CHZ868 is a potent JAK2-biased Type II inhibitor that has efficacy in pre-clinical models of MPN at high nanomolar concentrations (250-500 nM) but its effects on withdrawal signalling have not been studied. In contrast to Type I inhibitors, CHZ868 did not prevent JAK2 activation loop dephosphorylation as shown by in vitro phosphatase assay. CHZ868 was tested in SET2 cells and primary JAK2 V617F patient samples and did not induce accumulation of JAK2 phosphorylation. Most importantly CHZ868 did not exhibit rebound JAK2 signaling upon washout and was superior in the eradication of JAK2 mutant positive progenitors purified from 14 myelofibrosis patient samples. Immunophenotypically, CD34+ progenitor cells from myelofibrosis patients most resembled CD34+CD38+CD45RA+CD123hi granulocyte-myeloid progenitors and lymphoid-restricted multipotential progenitors.
Consistent with our signaling, chi-square test of 10 published cases of severe ruxolitinib-withdrawal syndrome noted a high frequency of JAK2 mutant cases (100%) compared to expected mutation frequencies (2 x 2 contingency table, P= 0.0203). In majority of cases, symptoms began within 72 hours.
Conclusions: Type I inhibitor-induced loop phosphorylation acts as a pathogenic signaling node released upon drug withdrawal, especially in JAK2V617F patients. Ruxolitinib-withdrawal syndrome may be more frequent in JAK2 V617F mutant cases.
Hughes:Incyte: Honoraria, Membership on an entity's Board of Directors or advisory committees; Novartis: Honoraria, Membership on an entity's Board of Directors or advisory committees; BMS: Honoraria, Membership on an entity's Board of Directors or advisory committees. Ross:Celgene: Research Funding; Novartis: Consultancy, Honoraria, Research Funding, Speakers Bureau; BMS: Honoraria.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal